Breast cancer risks associated with missense variants in breast cancer susceptibility genes
TL;DR: Helix has been used as part of the BRIDGES project to predict the impact of variants of interest on cancer susceptibility in a recent paper: Breast cancer risks associated with missense variants in breast cancer susceptibility genes.
Helix outperforms all competing predictors in 3 out of 4 novel datasets.
Introduction
Genetic testing for cancer susceptibility is now part of mainstream clinical practice. For breast cancer susceptibility, genetic testing generally focuses on high-risk genes (BRCA1, BRCA2, PALB2 and TP53). Testing of larger panels that include so-called “moderate-risk” genes is being increasingly offered. In a recent large-scale study performed by Dorling et al. a panel of 34 putative susceptibility genes was used to perform sequencing on samples from 60,466 women with breast cancer and 53,461 controls. Protein-truncating variants in 5 genes (ATM, BRCA1, BRCA2, CHEK2, and PALB2) were clearly associated with increased risk of breast cancer.
Besides PTVs, genetic testing also identifies missense variants for which the impact on protein function and associated cancer risk is generally unknown (“variants of uncertain significance”, or VUS). The large number and rarity of these VUSes presents a formidable obstacle as the interpretation of these variants is a difficult and time-consuming task and solid statistical analyses are often intractable.
In-silico predictions
Computational approaches that try to distinguish benign from pathogenic variants are routinely used, although these tools leave room for improvement, especially in a clinical context. In a follow-up paper by Dorling et al. the effectiveness of five in-silico algorithms in predicting breast cancer risk associated with missense variants in ATM, BRCA1, BRCA2, CHEK2 and PALB2 was studied.
Based on the same sequencing of samples from women with breast cancer and controls, logistic regression (LR) was used to explore which of the five in silico scores (Align-GVGD, BayesDel, CADD, Helix and REVEL - all analyzed as continuous variables) were most strongly associated with risk of breast cancer, analyzing rare missense variants in ATM (1,146 training variants), BRCA1 (644), BRCA2 (1,425), CHEK2 (325) and PALB2 (472).
For each of the five genes, logistic regression models were created using samples from the training data sets. In these models an in-silico predictor score is the independent variable and the case/control status is used as the dependent variable. When testing the models on the validation sets the models were very robust, with no (significant) difference in performance.
The most predictive in silico algorithms were Helix (BRCA1, BRCA2 and CHEK2) and CADD (ATM). There was little evidence for an association with risk for missense variants in PALB2. The figures below show the p-values for association for the five in-silico predictors for the genes ATM, BRCA1, BRCA2 and CHEK2. The negative log of the p-value is displayed, so higher is better.
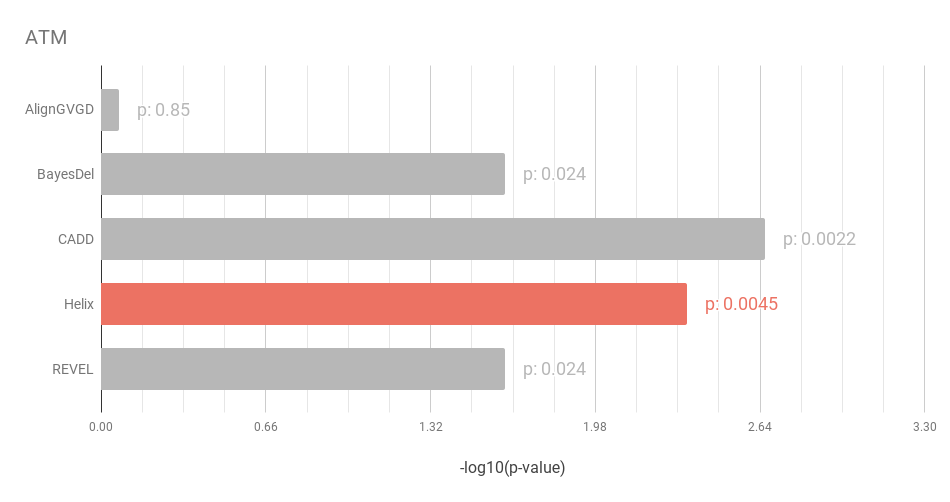 Figure 1: CADD performs slightly better than Helix on the ATM dataset.
Figure 1: CADD performs slightly better than Helix on the ATM dataset.
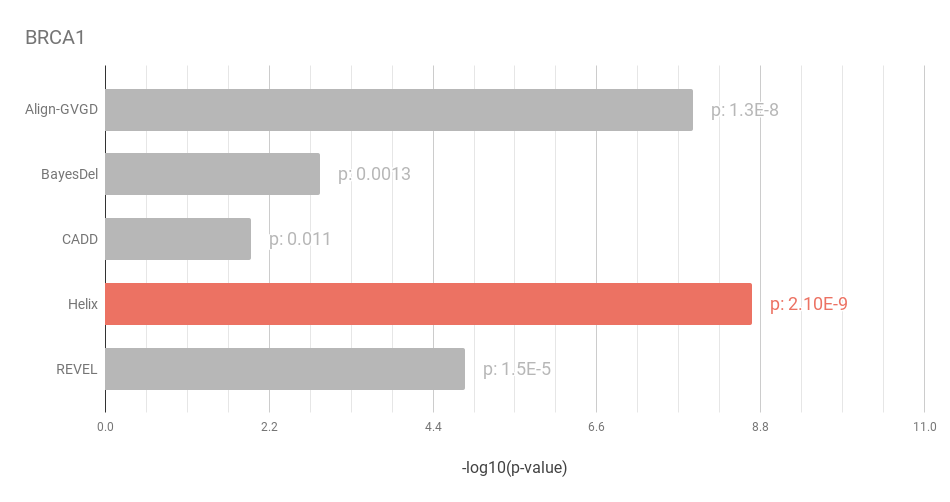 Figure 2: When evalutating BRCA1 predictions, Helix outperforms all other predictors.
This matches our observations on a separate dataset in the Helix white paper.
Figure 2: When evalutating BRCA1 predictions, Helix outperforms all other predictors.
This matches our observations on a separate dataset in the Helix white paper.
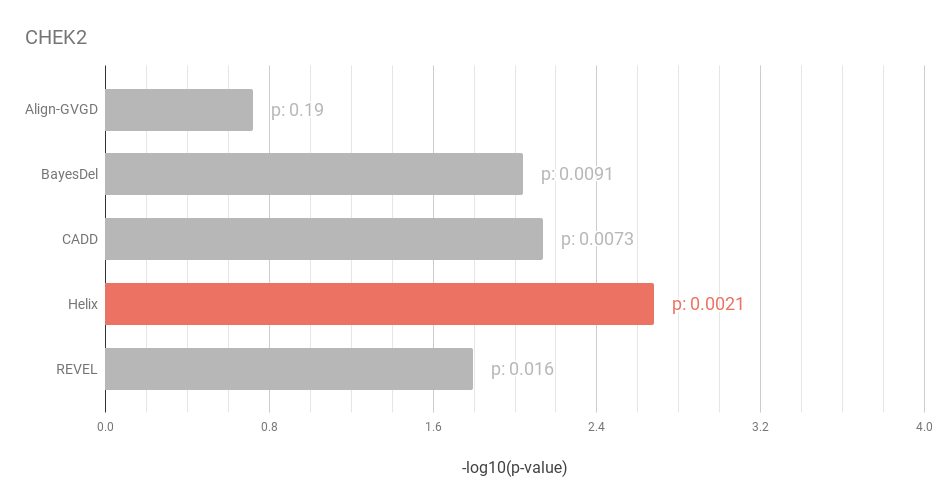 Figure 3: Cancer susceptibility caused by CHEK2 mutations is best predicted by Helix.
Figure 3: Cancer susceptibility caused by CHEK2 mutations is best predicted by Helix.
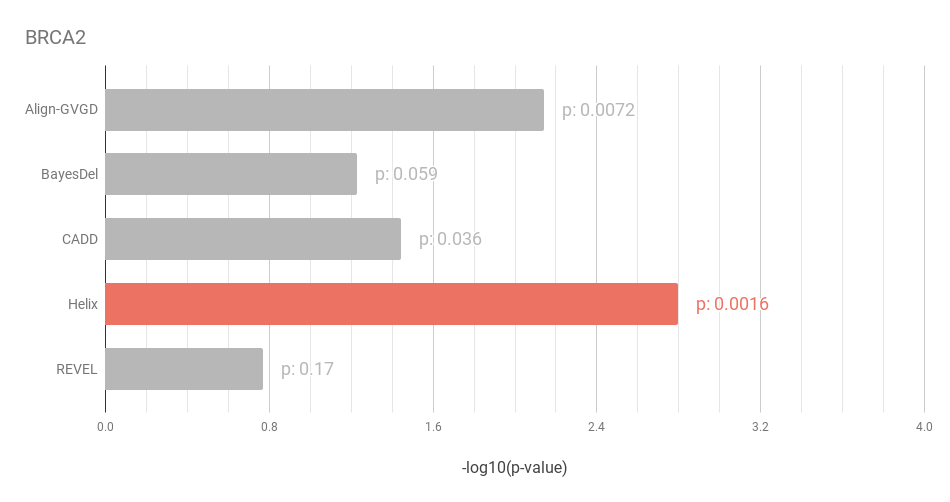 Figure 4: Similar to the results in the BRCA1 dataset, Helix performs best when predicting
the effect of mutations in BRCA2.
Figure 4: Similar to the results in the BRCA1 dataset, Helix performs best when predicting
the effect of mutations in BRCA2.
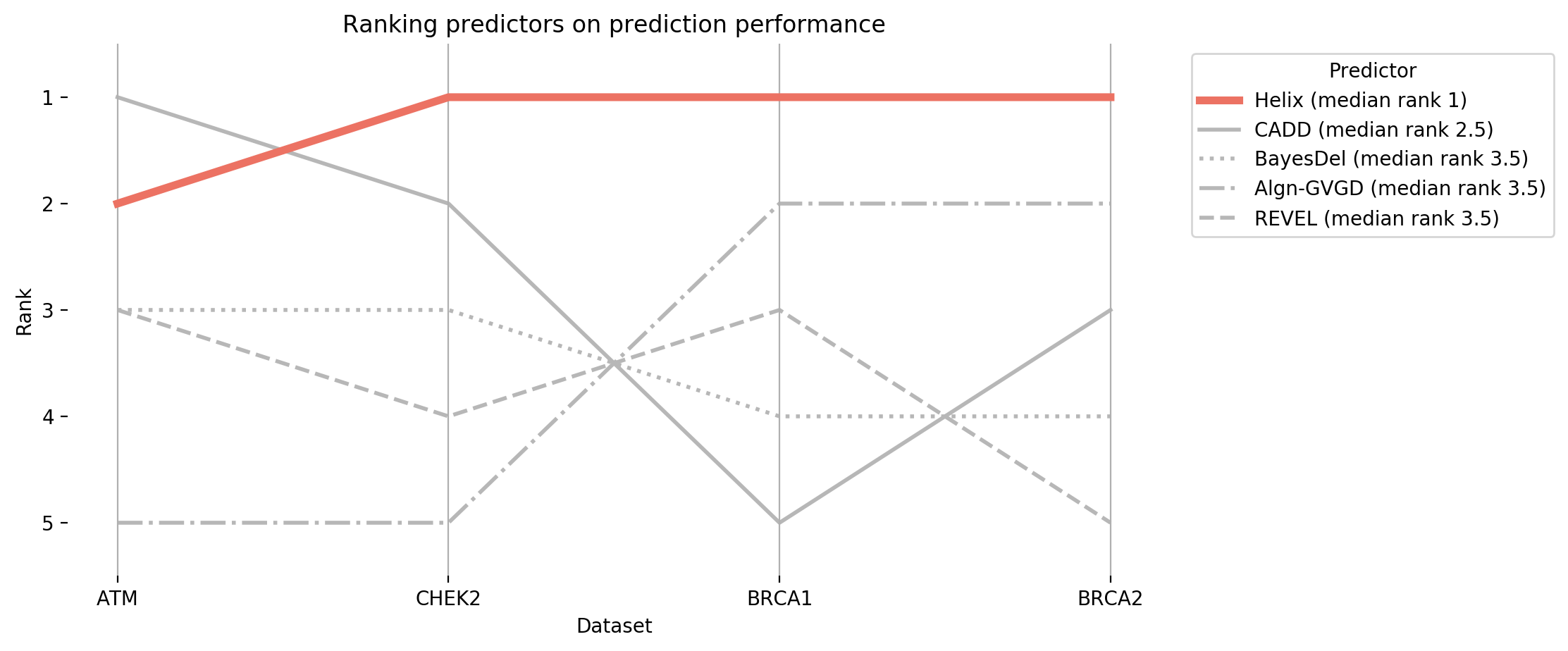
Figure 5: A comparison between the ranks of the 5 different predictors on these datasets.
Helix always scores highly, whereas other predictors are inconsistent in their ranking.
Functional assays
The authors also investigated whereas functional assays could be used to enhance risk association. Extensive functional data is available for BRCA1, and this data was used to augment in-silico predictions. The analysis of the functional assay scores for BRCA1 suggests that this approach should be useful, although the scores were highly concordant with the best in-silico score (Helix) in this case. The excellent performance of Helix on the functional assay BRCA1 data was already discussed extensively in our white paper.
Summary
In summary, this study confirms that subsets of missense variants in established breast cancer susceptibility genes are associated with increased risks of the disease and provides estimates of relative risks for those subsets, as well as probabilities for association with risk at the variant level. Helix is clearly the best performing in-silico predictor, where Helix predictions are most predictive of breast cancer risk. Since Helix was not specifically trained or optimized for breast cancer related genes, we expect the Helix will be similarly useful in other disease domains.