Frequently Asked Questions
- The website contains in depth variant reports, with literature references and explanations on the prediction made by Helix. Samples can be viewed in our portfolio.
- The API allows users to access Helix data in a structured manner, with full access to predictions for every missense variant in the human exome. Bulk methods are available for faster throughput.
- The flat file contains an overview of predictions and is suitable for loading into your in-house database. Restrictions apply.
For specific purposes one may prefer a focus on either precision or recall. We encourage you to contact us with your use case, we will supply you with an in-depth analysis of the best cutoff for you.
MCC (Matthew's Correlation Coefficient) is a metric that measures the quality of binary predictions. Pathogenicity datasets have binary labels (pathogenic or benign). ROC AUC (Area Under the Curve, specifically the Receiver Operating Characteristic curve) also measures binary prediction performance, but does so for every possible cutoff point.
ROC AUC is a valuable measure if being able to tune the cutoff for prediction is trivial. However, due to the variety of different genes with different properties, we find that it is difficult to do so in practice. We thus assess MCC in order to evaluate the performance of the predictor with the cutoff set to 0.5.
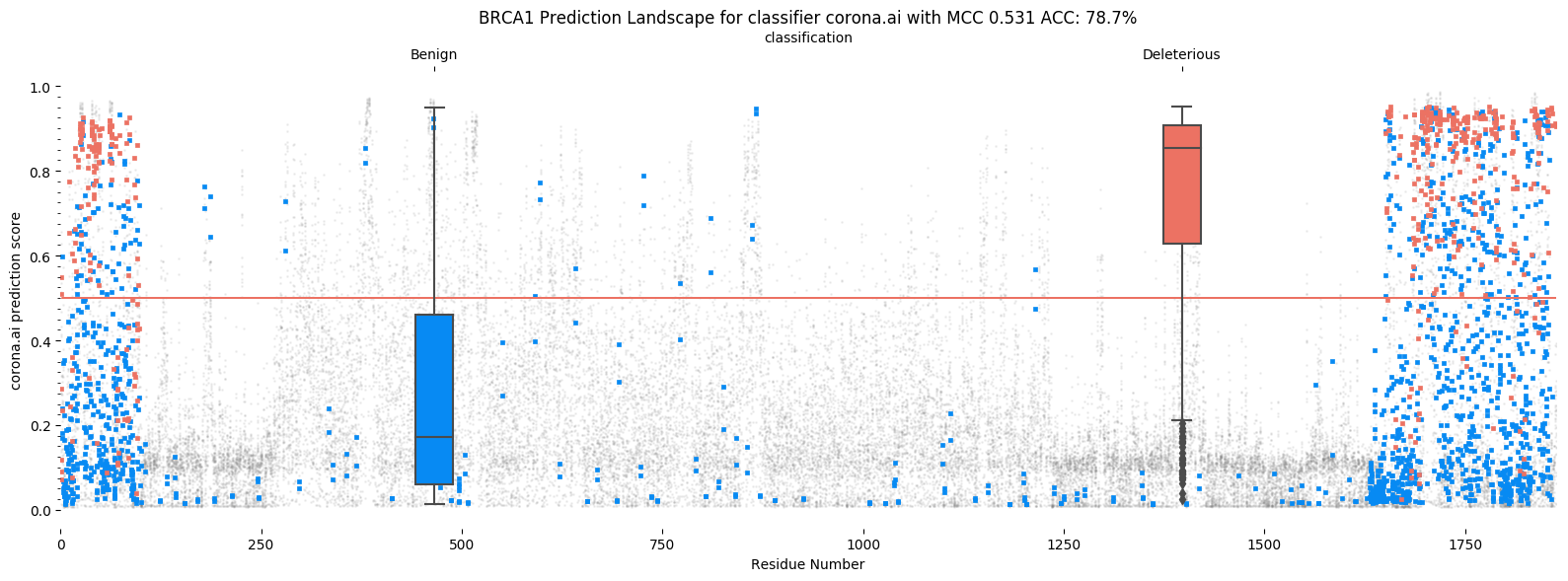
Illustrated below are two prediction landscapes. The X axis corresponds to the residue number of BRCA1,
while the Y axis shows the prediction value for Helix (v4.0.1) and REVEL. Red points indicate pathogenic variants,
blue points indicate benign variants. Grey points are variants of unknown significance.
In the middle of both plots sits a red line, indicating the 0.5 cutoff. A perfect predictor would place
all red points above the line and all blue dots below the line. Box-plots show the distributions of predictions
for each category.
Helix gene landscape
REVEL gene landscape
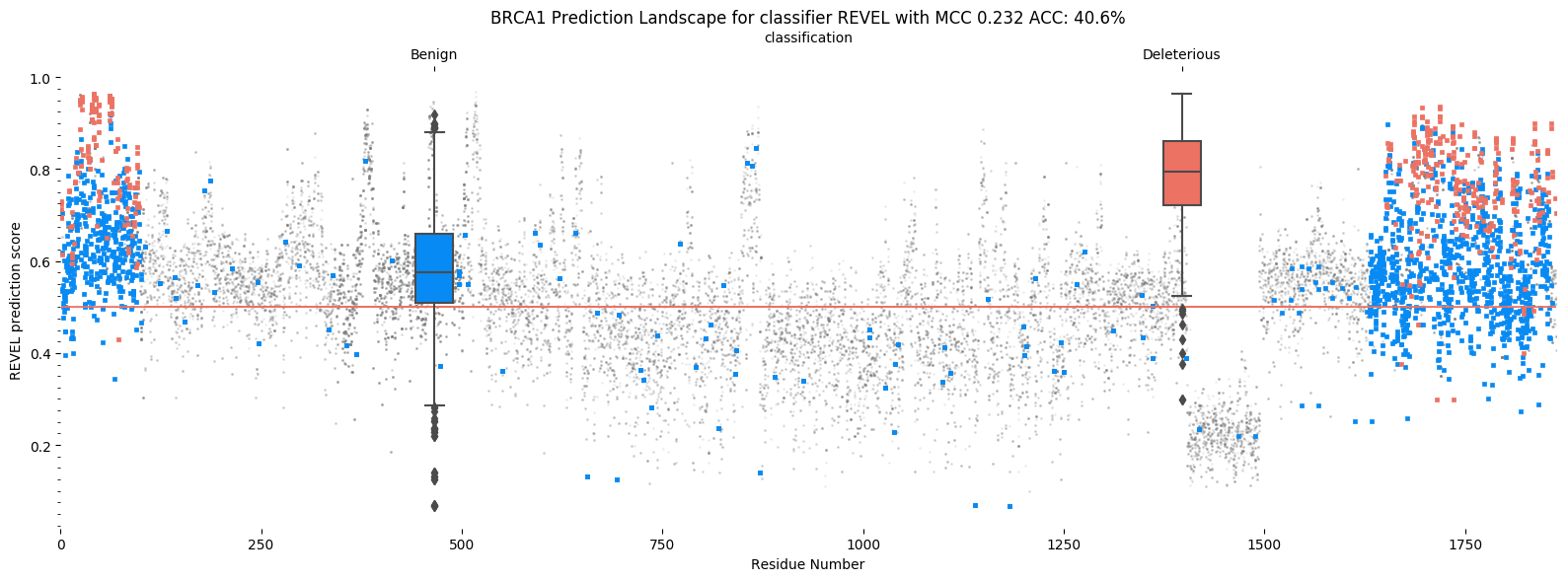
REVEL places the vast majority of points above the red line, predicting almost all as being pathogenic. Helix distributes them more evenly (though not perfectly, BRCA1 is a notoriously difficult gene to predict). This results in a better accuracy and better MCC for Helix.
It is possible to move the cutoff for REVEL in this case to produce a better MCC or accuracy score. This is roughly what ROC AUC measures. While this is possible for BRCA1, which is well studied, most genes are not as well characterized. Less than 10% of all human genes are associated with more than 10 known variants, most of these are benign. We deem adjusting cutoffs based on sparse data an error prone exercise. In order to address this, we elect to evaluate predictors based on their recommended cutoffs using MCC.
- Helix is available for every variant in the Human exome.
- Helix provides in-depth variant reports with access to literature for every variant.
- Helix is consistently updated, in contrast to other predictors that are frequently abandoned once the research project is finished.
- You can always contact the Helix team for information and requests related to predictions - and expect a timely response.
- Helix predictions are constantly improving, as novel techniques are incorporated into our predictor.
Is your question not listed? Don't hestitate to contact us!