White paper: The Helix Pathogenicity Prediction Platform
This is an HTML reproduction of the white paper hosted on ArXiv.
Introduction
Millions of human genomes and exomes have been sequenced, but their clinical application remains limited due to the difficulty of distinguishing rare disease-causing variants from benign genetic variation. Without clear lines of evidence that show that a missense variant is either benign or neutral, variants are deemed 'variants of uncertain significance', or VUS. The large number and rarity of these VUSes presents a formidable obstacle, as the interpretation of these variants is a difficult and time-consuming task, thereby severely limiting the actionability of diagnostic sequencing. Furthermore, the large amount of VUSes hinder the effective implementation of applications that require large amounts of genotype-phenotype associations such as disease association studies and patient stratification in drug development programs.
To deal with this ever-growing problem, a large number of computational tools have been developed that aim to predict the pathogenicity of novel variants. These tools are typically based on limited data describing evolutionary conservation and constraints, sometimes combined with the physicochemical characteristics of the amino acids and information about the structural domains in which these residues reside. The emergence of many 'meta-predictors' (or 'ensemble predictors'; predictors that take predictions of other predictors as inputs) has led to an increase in overall predictive performance. However, despite obvious progress, these tools leave room for improvement, especially in a clinical context.
At the core of missense variant prediction lies the problem of understanding the role of proteins and their constituent amino acids. A good understanding of this relation between sequence, structure and function is the ultimate goal that enables insight in the impact of missense variants.
Recently, advances have been made in closing this sequence-function gap by artificial intelligence (AI) methods, where contextual language models have been developed that can provide information about how proteins are formed, shaped and function based on protein sequences alone (Elnagger, 2020). Trained on billions of protein sequences, these models contain a wealth of information and can be applied to a wide range of predictive tasks with just one protein sequence as input. However, the predictive power of models solely based on these protein representations on explicit protein prediction tasks still fall short of predictive methods that include evolutionary information in the form of multiple sequence alignments (MSAs) or information derived thereof.
This evolutionary information (e.g., amino acid conservation patterns) can be used because functional and structural requirements lead to specific selective pressures on proteins. In turn, identifying these evolutionary constraints provides data and insights that can be used for missense variant effect prediction and interpretation.
The ability to detect and interpret these evolutionary signals is dependent on the quality of the data that is used. The application of deep and high-quality multiple sequence alignments integrated with protein structure data is commonplace in a number of fields that require detailed understanding of the role of individual amino acids. Protein optimization pipelines and drug development programs often critically depend on the availability of deep annotations and integration of protein sequence and structure data. Such high-quality protein annotation data has not been available at scale, and the lack of such data at large scale has hampered its application for missense effect prediction.
We present Helix, a missense variant effect predictor built on a resource of deep integrated protein data, in which over 30,000 protein family structure-based MSAs, over 50,000 protein structures and $\sim$60,000 human targeted sequence-based MSAs are combined to provide deep annotations for all human proteins at the level of individual amino acids. Helix incorporates this data in combination with Contextual Language Model representations that contain implicit information about how proteins are formed, shaped and function.
The features used by Helix describe proteins and their constraints at multiple levels. This includes extensive MSA-derived metrics describing selective pressures that act on proteins as a whole, on networks of residues required for specific functions and on individual positions. This information is integrated with features describing structural aspects, e.g. secondary structure, flexibility and solvent-accessible surfaces, both at the level of individual protein structures, as well as in the form of aggregated data across protein domains and families. Additionally, Helix uses descriptors of gene mutation tolerance, gene occurrence throughout the evolutionary tree as well as data describing protein-protein interactions. Best in class classification systems trained on this data are combined with state of the art AI methodologies to produce a predictor that offers the best of both worlds.
Helix was trained on a large set of well-annotated variants, using a strict training regime where circularity is actively avoided. We benchmark the performance of Helix using 10-fold cross validation datasets and two previously published, clinically relevant datasets. We show that Helix consistently outperforms existing predictors.
Predictions, however, are just that. For the effective use of these predictions more context is often required. To this end, Helix provides high-quality predictions together with detailed variant reports that contain references to relevant scientific literature, information about evolutionary constraints, underlying data quality assessment and interactive protein structures.
Methods
Helix is built on top of the proprietary 3DM platform. 3DM is a protein data and analytics platform that collects, combines and integrates protein data for protein (super-)families. Examples of data included for every protein family are protein sequences and structures, SNP data from whole genome sequencing studies, scientific literature and patents, and data derived thereof. Built around uniquely deep and high quality structure based multiple sequence alignments, the 3DM platform offers data and tooling that enable researchers to efficiently investigate the relation between protein sequence, structure and function. Details about the methodology behind 3DM can be found in Kuipers et al.(Kuipers, 2010).
Currently there are over 30.000 3DM protein family systems available. These are built using templates selected from a) the SCOP classifications of protein structures, and b) by clustering the structures in the PDB. Overall, these cover the complete structural space, and serve as an extremely rich data source with which to annotate all human protein sequences. In short, this resource contains high-quality MSA data integrated with protein structure data, scientific literature, SNP data, etc for all parts of sequence space that can be mapped to an existing protein structure. In terms of human proteins, this resource can be used to annotate any part of a human protein that can be aligned with a (not necessarily human) protein structure.
Annotating the human proteome
To enable predictions for the complete human proteome, it is necessary that every individual amino acid is annotated. Since there are many (parts of) proteins for which no structure information is available and therefore cannot be annotated with the data present in the 3DM platform, full-length sequence based alignments are a necessity to ensure full sequence coverage. Therefore, Helix bases predictions not just on a single alignment - where possible multiple alignments with different depths are used to provide optimal predictive power. All positions in the human exome are covered by full length, sequence-based multiple sequence alignments (MSAs). In addition, (PFAM) domains are extracted and covered by deeper, more specific alignments. In areas for which a mapping to a protein structure can be made, the deep structure-based alignments (and associated data) from the 3DM platform are used. In typical sequence based MSAs there is a very distinct tradeoff between coverage, alignment depth and alignment quality. In contrast, by using multiple MSAs the requirement for full sequence coverage is fulfilled, while maximising alignment quality and depth at the same time.
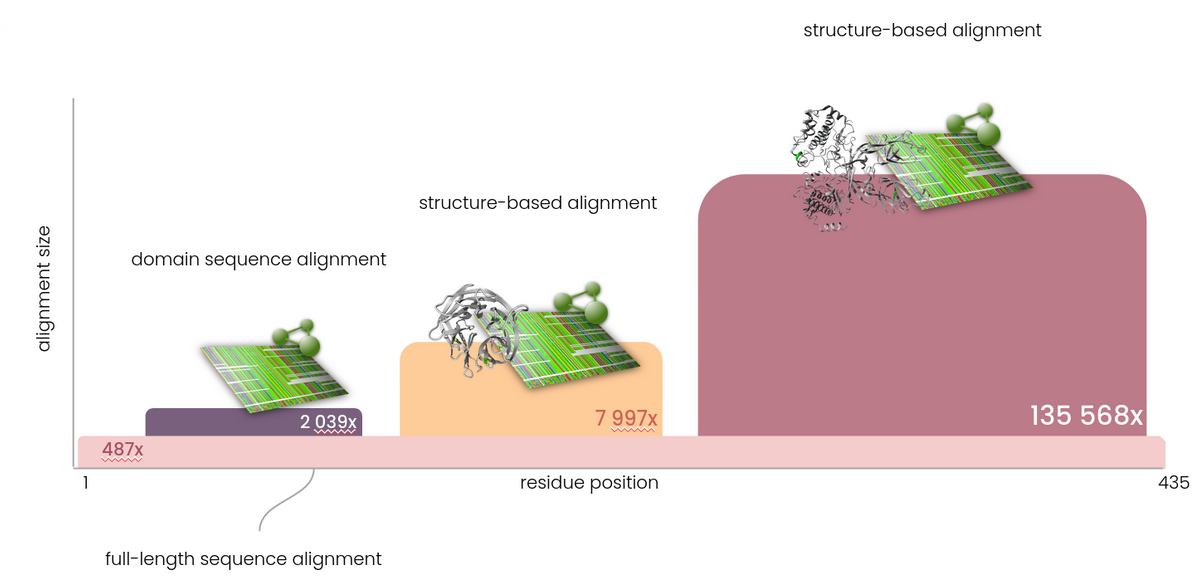
Features
The features used in our tabular data models are hand curated and include a large number of descriptors of evolutionary pressure that are extracted from multiple sequence alignments. These include several indicators of evolutionary pressure, acting on proteins as a whole, on networks of residues required for specific functions and on individual positions. Metrics include measures of conservation and change, entropy metrics, correlated behaviour and where available, extensive structural data (e.g. solvent exposure, electrostatic interactions, hydrogen bonding) that describe the structural aspects of individual amino acids. The feature set is expanded with a set of indicators of vulnerability on the gene level. Minor allele frequency is explicitly not used as a feature, as this would introduce a large amount of circularity and predictive performance on rare variants would be very limited as a result. Predictions from other classifiers are not used as a feature, meaning that Helix is built from the ground up without suffering from restrictions on the training data.
Datasets and data preprocessing
The reference variant dataset used in this whitepaper was composed of variants from ClinVar (Landrum, 2018) and gnomAD and a dataset maintained by the Dutch genome diagnostic laboratories (VKGL). ClinVar variants were included if the Clinvar review status was one star or higher, excluding variants with conflicting interpretations. ClinVar variants with "Benign" and "Likely benign" interpretations were included and labeled as benign variants, whereas those variants with "Pathogenic" and "Likely pathogenic" interpretations were included and labeled as pathogenic variants. gnomAD variants were selected and labeled benign if the minor allele frequency exceeded 0.1%.
Machine learning
10-fold cross validation was used to train and analyze model performance. In this process, the training data is divided into 10 partitions, where repeatedly 9 partitions are used for training and one for validation. The details of how this partitioning of the data takes place has important consequences for model performance and generalizability to real-world applications.
Most practical implementations of the 10-fold cross validation technique use a random split, where the selection of variants that end up in the training or validation set for each fold is performed completely random, irrespective of gene or residue positions. As we have shown in previous research(Heijl & Vroling, 2020) this leads to issues with circularity and artificially increases validation scores. In order to mitigate inflated scores, we use a gene split strategy, where variants present in training and validation sets are separated by gene; i.e. all variants for any given gene are either present in the training set or in the validation set. Variants present in the test sets (BRCA1(Findlay, 2020), Clinical (Gunning, 2020)) were excluded from the training/validation data and exclusively used for testing.
Ensemble prediction
Using a variety of models to predict pathogenicity and combining their predictions into an ensemble allows for better predictive performance than using a single classifier(Opitz, 1999). This process is used to combine all our models, while restricting the training and test set combination to prohibit any contamination between datasets.
Models used in the ensemble prediction include state of the art Gradient Boosting classifiers, custom neural networks that perform well on tabular data and proprietary contextual language models. The latter are trained on a multitude of subtasks to infuse the model with external data related to the pathogenicity prediction problem.
Results
The primary metric that was used for performance assessment is Matthew's Correlation Coefficient. MCC is robust in the face of imbalanced datasets and will report low scores for naive results. Comparisons are made with a number of commonly used baseline, as well as newer predictors. The former includes SIFT (SIFT, 2019), CADD (Rentschz, 2019) and PolyPhen2 (Adzhubei 2010), while the latter consists of REVEL (Ioannidis, 2016) and VEST4 (Carter, 2013).
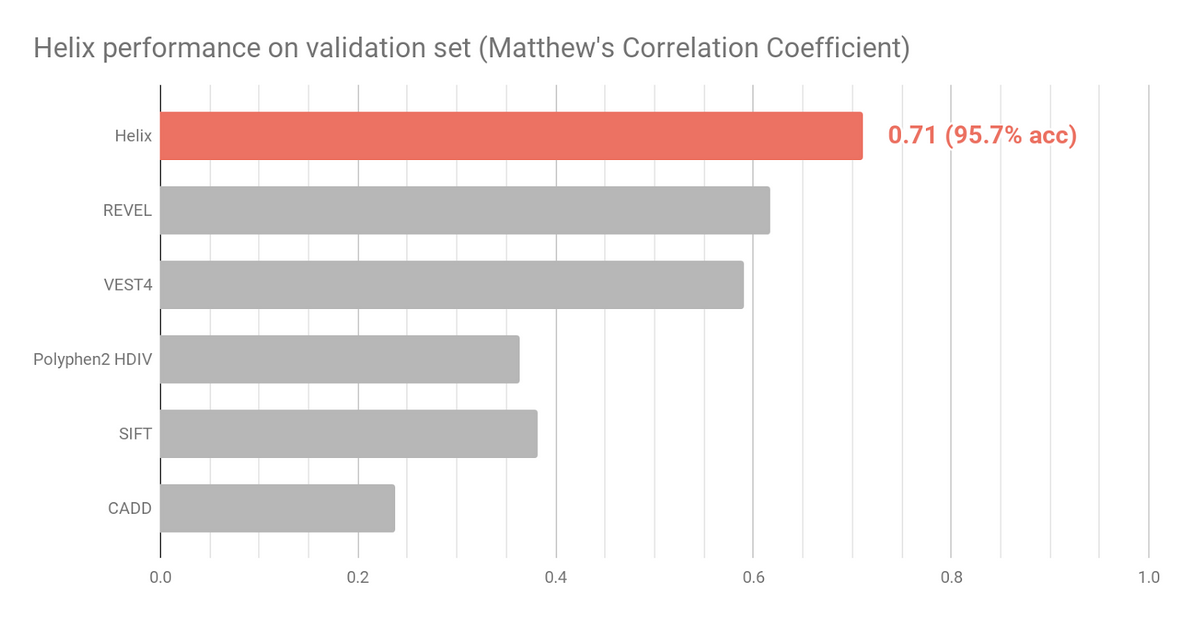
Figure 2 shows the Helix performance on the validation set. Helix predictions were obtained by using 10-fold cross validation. Prediction scores were obtained by testing on unseen genes, a more stringent way of testing compared to the evaluation of the other tools listed. To minimize training bias effects and ensure an as fair as possible comparison, variants were excluded from this analysis if present in the HGMD (Stenson, 2009) dataset or the PolyPhen HumDiv andHumVar training sets.
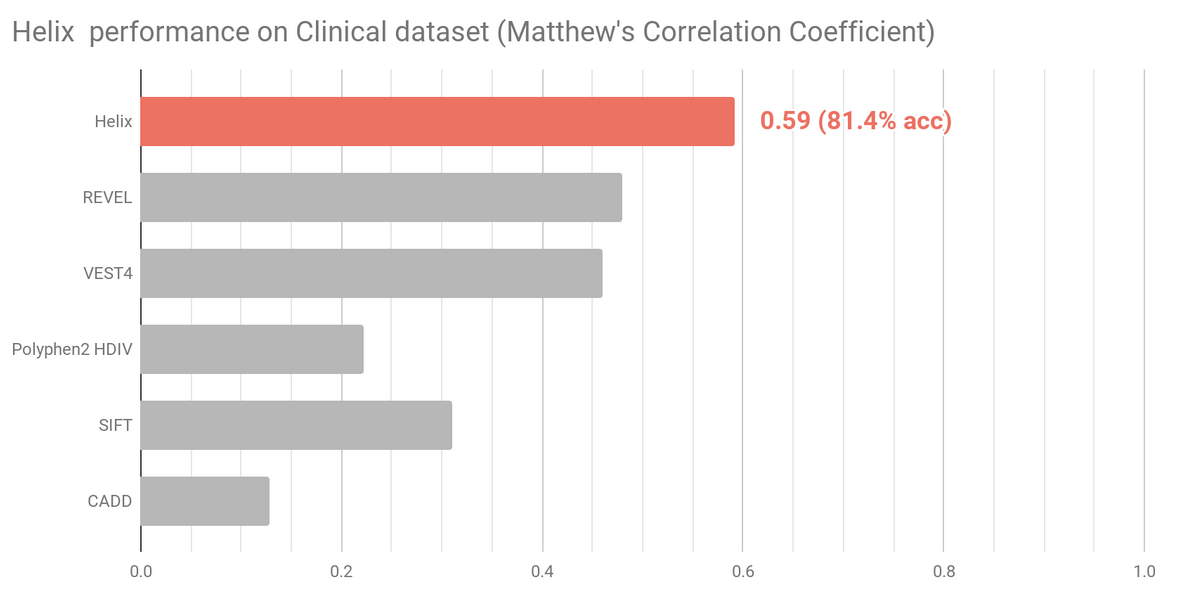
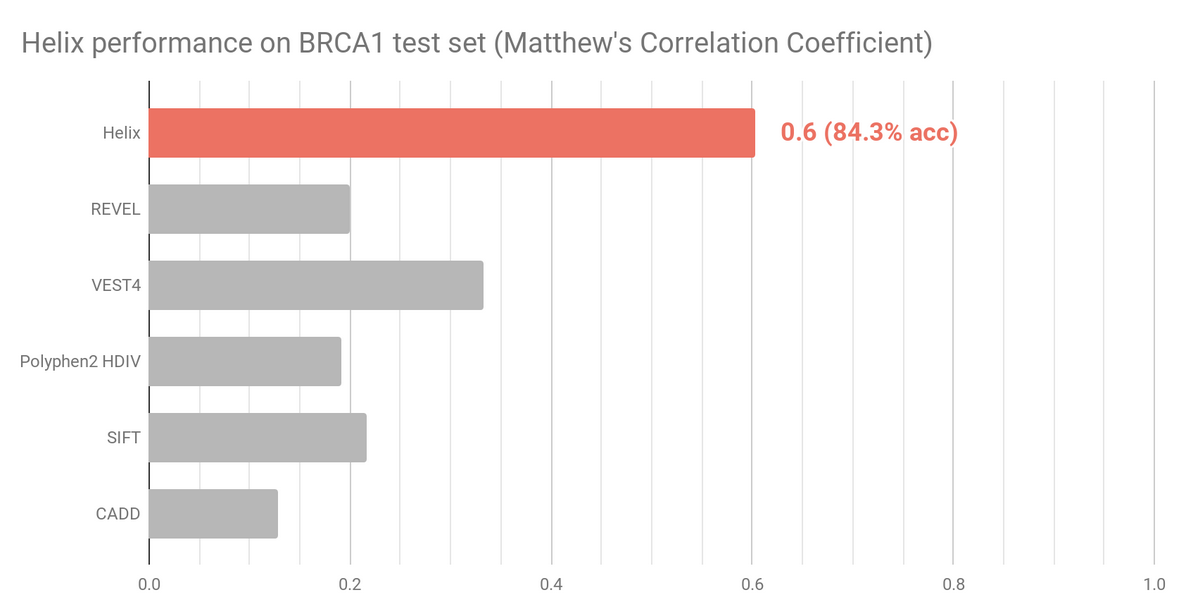
Two datasets that more accurately reflect variants that might require classification in a clinical setting were selected to independently assess and compare Helix predictions (Figure 3 and Figure 4). Variants present in these sets were excluded from the Helix training data. Overall, these datasets are composed of variants that are clinically relevant, have been reviewed recently, and where there is very limited overlap with training datasets of existing predictive tools.
The Clinical dataset (n=1729) contains variants from the Deciphering Developmental Disorders (DDD) study together with manually classified clinical variants and variants identified in Amish individuals. Figure shows
The BRCA1 dataset (n=1605) contains experimentally assayed BRCA1 variants using saturation genome editing.
Reports
Helix predictions can be accessed through the web application, where reports for every single variant in the human exome can be found. Reports annotate variants with structural information, literature and prediction data, with the ultimate goal of providing domain experts the data and information they need to make the best decisions.
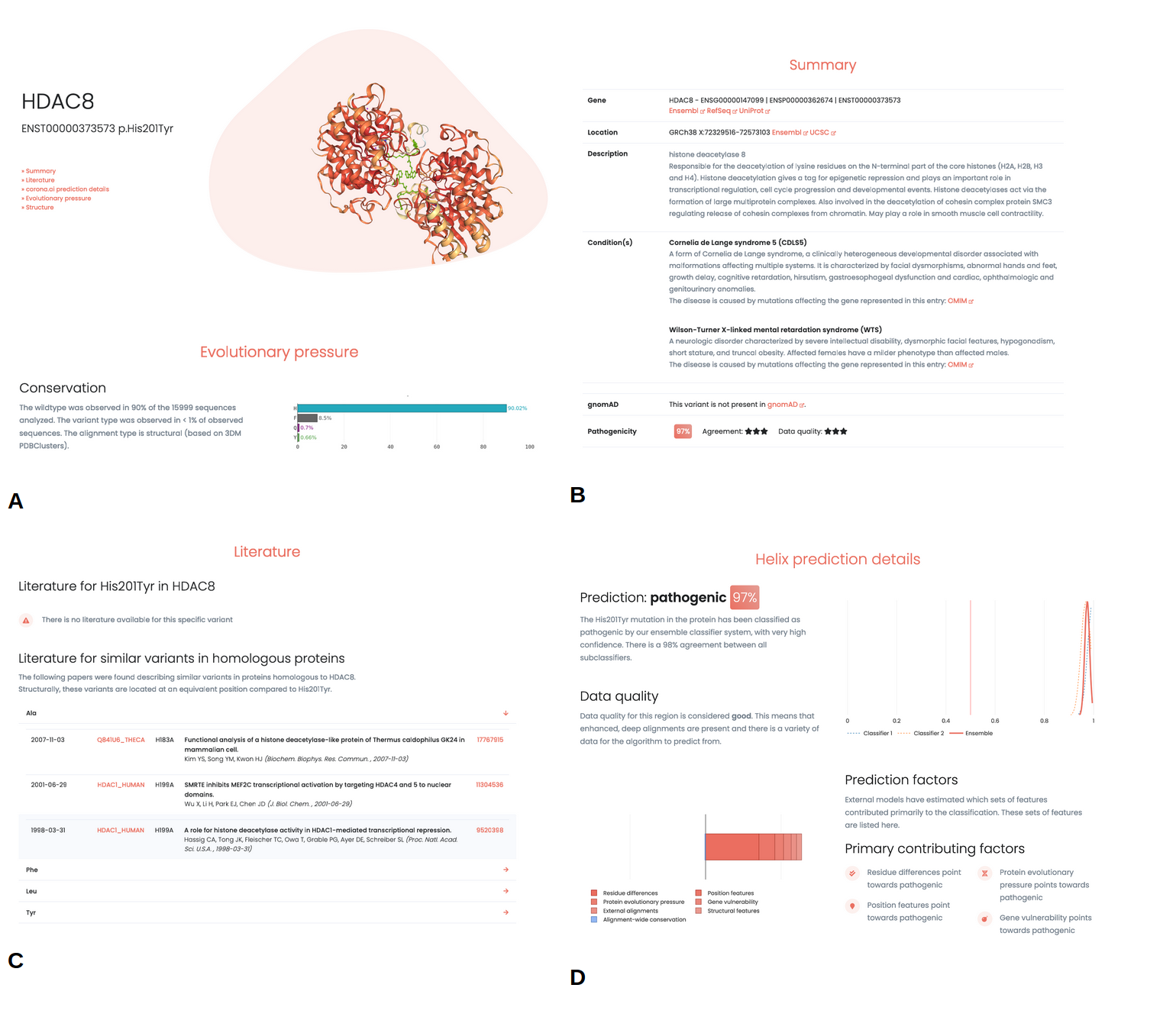
Variant information
WIthin the reports information is presented about the individual variant. Known associated phenotypes for the gene are described, together with relevant information about the affected gene and links to external sources (e.g. ClinVar, gnomAD, UCSC, Ensembl). Furthermore, information about the conservation for the wildtype and variant type residue is presented, together with structural information (Figure 5 A, B). Literature associated with the variant is listed. Importantly, this is not limited to papers describing the exact variant but extends to variants described in homologous proteins (Figure 5, C).
Prediction
The variant report provides details on various aspects of the predictions (Figure 5, D). Agreement between the different sub-predictors in the ensemble is shown, where a high agreement generally indicates more certainty in the prediction.
A diverse set of metrics is used to present an intuitive score indicating the quality of the data underlying the prediction. This score is an indication of the amount of confidence one should place on a given prediction. No matter how good the predictive model is, if the data that is provided to the model is of limited quality (i.e. small MSAs, no protein structures available), one should be careful in placing too much confidence in these predictions.
In addition, the report offers a visual indication of the factors that are estimated to contribute most to the individual predictions. Features are grouped into a few categories to simplify interpretation.
Discussion
Helix is built on a unique resource of integrated protein data, and represents a state-of-the-art predictive tool that outperforms currently available predictors by a large margin. However, Helix is by no means 'finished'; many challenges remain open for the problem of predicting the effects of missense variants. In fact, despite offering impressive predictive performance, Helix is just getting started. Novel challenges lie in problem domains such as accurate modeling of protein-protein interactions and the integration of pathway information to be able to assess downstream effects of variants within the context of larger systems.
Helix offers an interactive platform that allows researchers to investigate both the variants as the predictions in great detail. The variant reports, in which variants and their associated predictions are presented in a coherent fashion to add context and interpretability, offer an important first step to help researchers make sense of all that data.
In the future, Helix will continue to integrate novel technologies and performance increasing features. Helix is supported by a longstanding bioinformatics company that is dedicated to its further development. As the AI field continues to provide new solutions that touch the variant effect prediction space, we envision including these technologies with even better performance and interpretability as a result.
Acknowledgments
The research leading to these results has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 634935 (BRIDGES), No 635595 (CarbaZymes) and No 685778 (VirusX).